

Early Marketing Authorization and Market Access
The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have recognised that paradigms of drug development, which are feasible for common diseases, may not be feasible for rare diseases and that transformative orphan, specialty and advanced therapeutics often defy traditional regulatory routes.
The EMA’s Support for Early Access and the FDA’s Expedited Programs aim to facilitate and accelerate development and marketing authorisation. The programs a focus on life-threatening and debilitating diseases with a major impact on quality of life (defined as serious conditions), and on medicines with a credible promise of significant improvements in clinical benefit and patient-relevant outcome(s).
While the FDA’s programs are well established, certain of the EMA’s early access tools have been launched more recently. These innovative initiatives emphasise early dialogues, the involvement of multiple stakeholders, iterative development in a life-cycle approach, and an expanded toolbox for evidence generation, with pragmatic and real-world studies complementing RCTs in areas where the collection of data via traditional routes is difficult. Early access does not alter the current FDA or EMA standards of approval.
There is substantial crossover in terms of objectives and features between the programs and they can be used in combination. For example, a product eligible for fast track in the US may be eligible for accelerated approval and priority review. In Europe, a medicine benefitting from PRIME support may also qualify for conditional marketing authorisation. Additionally, drugs benefitting from early access are those eligible to the EMA centralised procedure and a single marketing authorisation.
Early access is applicable to both orphan and non-orphan medicines in both jurisdictions, although the programs are more accepted for orphan drugs. Orphan drug designation is not an early access tool per se, and orphan medicines do not automatically qualify for accelerated procedures. Nevertheless, orphan drugs are highly likely to be eligible for early access. Therefore, the feasibility of orphan designation should be considered as part of any early access strategy, and equally, the potential benefits of early access should be considered as part of the decision to seek orphan designation.
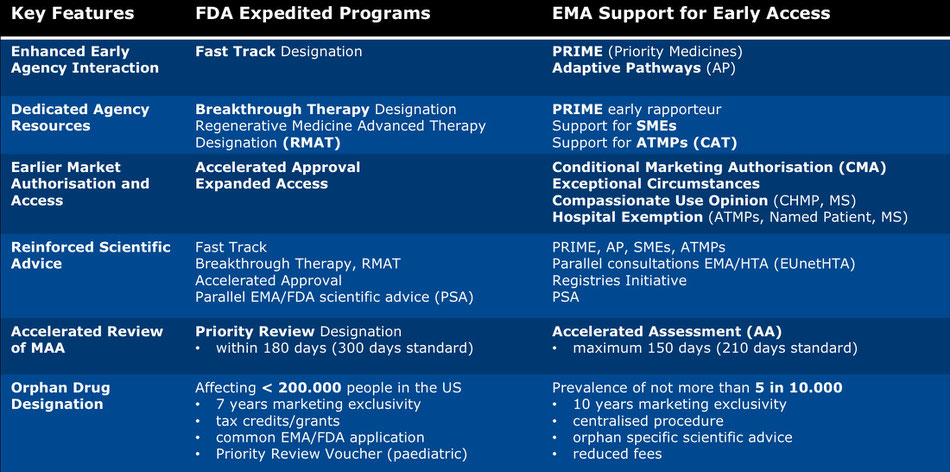
The above table provides an overview of the EMA’s and the FDA’s early access programs and represents the current regulatory context within which an early access strategy for an innovative medicine is developed. More on early access strategies can be found here:
The following is a full listing of the current early access programs, legal tools and initiatives in Europe and the United States. The blue highlighted external links to the EMA and FDA websites are current as accessed in February 2019 (note that ORPHA Strategy is not responsible for the content of the external sites).
European Medicines Agency (EMA)
Accelerated Assessment (AA)
- Reduces the timeframe for review of an application for marketing authorisation
Conditional Marketing Authorisation (CMA)
- Earlier authorisation of medicines fulfilling a positive benefit/risk balance for patients with unmet medical needs, on the basis of less complete clinical data, and where the benefits of immediate availability outweigh the risks that additional data are still required.
- Subject to specific obligations for comprehensive clinical and real world evidence to be generated within an agreed timeframe; valid one year, renewable.
- EMA Report - CMA 10 Years of Experience at the EMA
PRIority MEdicines (PRIME)
- Early, proactive and reinforced scientific, joint regulatory and HTA support at key milestones in development to identify potential for accelerated assessment.
- Access to dedicated EMA resources and early rapporteur appointment.
- A quick 2-page EMA primer on PRIME - Paving the way for promising medicines for patients
- Two Years of PRIME - update report
Adaptive Pathways (AP) and Early Dialogues with EMA and Health Technology Assessment Bodies
- A prospectively planned, iterative scientific development concept in areas where collection of data via traditional routes is difficult (e.g. rare diseases).
- Permits a stepwise approval in tightly defined patient populations with a gradual extension of the target population as more data become available. The standards of regulatory approval remain unchanged.
- Involves collaborating with all relevant stakeholders very early in the development process, particularly with HTA bodies and patients.
- “Safe harbour”, informal, non-committal entry to explore alternative development routes.
- Expands the toolbox for evidence generation, with pragmatic and real-world studies complementing RCTs.
- Download: Questions and answers following the initial experience of the Adaptive Licensing Pilot project
- Adaptive Pathways Workshop, December 2016, EMA documentation and a summary from the industry perspective: ORPHA Executive Briefings
- Early Dialogues Working Party (EDWP) - EUnetHTA
Initiative for Registries – “late dialogues”
- Facilitate the use of existing and the establishment of new registries to generate refined real-world based benefit/risk and value assessments
- Informal, collaborative approach for protocol design that utilises EMA resources and collaborations and will not interfere in the regulatory process
- Opportunity for interactions across committees and regulatory bodies (CHMP, PRAC, COMP, SAWP, HTAs)
- Further information in the report of the EMA patient registries workshop
- Report on CAR T-cell therapy Registries Workshop 9 February 2018
- Haemophilia Registries Workshop, June 2018
Orphan Designation for Medicines for Rare Diseases
- To qualify for orphan designation, a medicine must meet a number of criteria: it is being developed for treatment, prevention or diagnosis of a life-threatening or chronically debilitating disease, the prevalence of which in the EU must not be more than 5 in 10,000, and it must address an unmet medical need and/or be of significant benefit to patients.
- Medicines obtaining an orphan designation benefit from a number of incentives: 10 years market exclusivity, access to the centralised procedure, early access tools, scientific advice specific to designated orphan medicines, and reduced fees for regulatory procedures, SMEs receive additional incentives.
- Many products fulfilling the criteria for orphan designation will also qualify for early access. Therefore, the feasibility of orphan drug designation should be evaluated as part of any early access strategy, and, equally, the potential benefits of early access should be considered as part of a decision to seek orphan designation
Compassionate Use Opinion by the CHMP
- Facilitates early market access through a centralised compassionate use opinion by the CHMP, for unauthorised products, aimed at harmonising the conditions of use, distribution and the target population across the EU.
- To benefit seriously ill patients that cannot be treated satisfactorily or that cannot enrol in ongoing clinical trials.
Parallel consultation with regulators and health technology assessment bodies (as of July 2017)
- Aims to allow medicine developers to obtain feedback from regulators and health technology assessment (HTA) bodies on their evidence-generation plans to support decision-making on marketing authorisation and reimbursement of new medicines at the same time.
- These consultations can take place before or after the product is made available on the market.
- The objective is to help generate optimal and robust evidence that satisfies the needs of both regulators and HTA bodies.
- Early Dialogues Working Party (EDWP) - EUnetHTA
Small- and Medium Sized Enterprise (SME) office and Innovation Task Force (ITF)
- Small- and medium sizes enterprises (e.g. biotechnology startups) as well as academic innovators receive enhanced administrative, regulatory, scientific and financial support from the EMA, particularly for development in rare diseases and orphan designations.
Patients' and Consumers' Working Party (PCWP)
- Early and systematic inclusion of real life patient experience and views regarding benefit/risk and value in regulatory and HTA output
Medicines Search the EMA Website
- European public assessment reports - allows you to find the European public assessment reports (EPAR) for human medicines published by the European Medicines Agency (EMA)
- Pending EC Decisions - find medicines that have been evaluated by the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use Committee for Medicinal Products for Human Use (CHMP) and are currently pending a decision by the European Commission.
- EMA - ODD - allows you to find information on rare disease (orphan) designations based on applications that have been assessed by the European Medicines Agency's (EMA) Committee for Orphan Medicinal Products (COMP).
EU Register of Medicinal Products
- Orphan and non-orphan products, active, withdrawn or expired and refused (alphabetical or by EU number)
US Food and Drug Administration (FDA)
Expedited Programs for Serious Conditions - Drugs and Biologics
- Fast track designation is designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need. It is most beneficial early in development and features frequent interactions with the FDA fast track team regarding study design, safety data, surrogate endpoints, accelerated approval, rolling review, and priority review.
- Breakthrough therapy designation is for products where preliminary clinical evidence indicates substantial improvement over available therapy in serious diseases. Its key feature is an FDA commitment of significant resources with the aim of rapid development, including access to FDA senior management and the assignment of a dedicated cross-disciplinary project lead.
- Accelerated approval is an alternate development pathway that allows drugs or serious conditions that fill an unmet medical need to be approved earlier based on a surrogate or intermediate clinical endpoint, with the subsequent requirement for post-marketing confirmatory trials to verify the predicted effect on morbidity or mortality or other clinical benefit.
- Priority Review designation directs attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the treatment, diagnosis, or prevention of serious conditions (6 months vs. 10 months standard review).
- guidance for Industry on Expedited Programs - download guidance document (PDF)
Office of Orphan Products Development (OOPD)
- Developing Products for Rare Diseases & Conditions: the OOPD's mission is to advance the evaluation and development of products that demonstrate promise for the diagnosis and/or treatment of rare diseases or conditions.
- Orphan Drug Designation - provides orphan status to drugs and biologics which are intended for rare diseases/disorders that affect fewer than 200,000 people in the U.S., or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing. Orphan designation incentives include a seven-year marketing exclusivity period and tax credits for clinical testing.
- Orphan Drug Designation - Webinar Tips for Creating an Orphan Drug Designation Application
- Rare diseases - common issues in drug development guidance (PDF)
- Search FDA Orphan Drug Designations and Approvals
Center for Biologics Evaluation and Research (CBER) - Cellular & Gene Therapy Products
- Cellular & Gene Therapy Products overview page
- Cellular & Gene Therapy Guidances
- Human Gene Therapy for Rare Diseases - Draft Guidance
- FDA Approved Cellular and Gene Therapy Products
- RMAT - Regenerative Medicine Advanced Therapy Designation
FDA's Application of Real World Evidence
- Real World Evidence overview page
- Framework for FDA's Real World Evidence Program, PDF, December 2018
- Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices, PDF, August 2017
- FDA’s MyStudies Application (App)
FDA further links, including FDA & EMA Collaborations
- Rare Diseases Cluster: the FDA and EMA have set up a new ‘cluster’ on rare diseases to share experiences and best practices on each other’s regulatory approach. Topics include trial design, endpoints, development support, real-world evidence study design, risk management strategy.
- The cluster has developed common procedures for applying for orphan designation and for submitting annual reports on the status of development of designated orphan medicines: common EMA/FDA application
- CDER Patient-Focused Drug Development (PFDD)
- Parallel scientific advice EMA/FDA (PSA): one of the focus areas are orphan drug
- Clinical Outcome Assessment Compendium - the pilot COA Compendium is part of FDA’s efforts to foster patient-focused drug development - Compendium PDF download
Expanded Access (Compassionate Use)
- Expanded access, sometimes called "compassionate use," is the use outside of a clinical trial of an investigational medical product (i.e., one that has not been approved by FDA).