
Selected Publications and Case Studies

The Speaker: David Schwicker, MA, Principal, ORPHA Strategy Consulting
The Moderator: Marek Babusiak PhD., Training & Development Specialist, Symmetric
The Issue
With several hundred clinical programs in gene and cell therapies ongoing, there is enormous potential to address the unmet needs of patients with rare and complex conditions, which currently have inadequate or no satisfactory treatment options. But not all of these programs will advance to the commercial stage, given the substantial challenges in meeting the regulatory requirements for clinical evidence as well as manufacturing. This webinar concentrates on the newest strategies to successfully design and execute gene and cell therapy clinical trials.
Key Topics
- Accelerating development from early phase to scale up and regulatory approval
- Reducing the time and cost of clinical development programs and thoughts on de-risking
- Designing Phase I/II studies that are robust enough for marketing application
- Developing endpoints for gene and cell therapy trials - the crucial role of natural history studies
- Successfully identifying and recruiting patients and conducting trials in small populations
- Leveraging real-world evidence to enrich populations and enable faster, shorter trials
- The long-term follow-up of patients and post-authorisation clinical and value evidence generation
Webinar Recording
Webinar Slides Download
Adaptive Pathways and Real-World Evidence: Three Perspectives
Originally published in Pharmaceutical Executive Online (PharmExec.com), March 20, 2018

The Panel: Three HealthCare Stakeholders, Three Diverse Perspectives
The Moderator: David Schwicker, MA, Principal, ORPHA Strategy Consulting
The Issue
With a healthcare landscape driven by the advent of precision medicine and empowered patients, it would seem that “science has overtaken the system”. This includes the recognition that patients are often willing to accept greater risks from treatment of life-threatening and severely debilitating diseases in return for earlier access, especially when there are no alternative treatments available. This underlines a “need for speed” in biomedical innovation: more rapid development, accelerated approval and – crucially – timely access to transformative medicines. These developments challenge established clinical development pathways and put pressure on regulatory and particularly HTA (health technology assessment) processes.
In response, paradigms of biomedical innovation are being transformed with “Adaptive pathways” or MAPPS (Medicines Adaptive Pathways to Patients), which encompass earlier cross-stakeholder engagement, including patients, early access regulatory tools, and iterative evidence generation through the life cycle of the medicinal product with an expanded toolbox: pragmatic and real-world studies complementing RCTs where the collection of data through traditional routes is difficult, e.g. in rare diseases and in increasingly fragmented populations.
In order to achieve equitable and timely access to transformative medicines for patients with unmet needs, rapid market access and reimbursement in step with earlier conditional marketing authorisation is just as essential. However, the uncertainty about whether the anticipated clinical benefit will be verified in clinical practice and the possibility of undiscovered risks pose serious challenges to payers and HTA bodies. With greater uncertainty, a novel medicine’s lifetime price should no longer be determined at first market entry for a limited population, but be governed by more flexible and adaptive approaches to pricing and reimbursement that reflect the emerging evidence generated.
Patient-centered RWE presents a significant opportunity to enhance early access regulatory decision-making as well as adaptive value-based contracting centred on mutually agreed outcomes. Technological advances permit the collection of RWD from an increasing number of sources and platforms, while refined observational methodology and next generation analytics enable the translation of these data into RWE. Nonetheless, opinions on RWE remain diverse. Questions raised by stakeholders concern the methodology, reliability and the usefulness of RWE in decision-making, particularly regarding treatment effects. Based on these criticisms, it seems that RWE still has some way to go before becoming established as a universally accepted complement to RCT data for benefit/risk and value decision-making.
At last year’s ISPOR 20th Annual European Congress in Glasgow, UK, a panel offering “three diverse perspectives” — patients, payers, and biopharma — debated how the spectrum of stakeholders can improve the process of demonstrating benefit/risk and value in the challenging environment of adaptive pathways with less data and greater uncertainty; the role of real-world evidence (RWE) to support conditional authorization and early market access; the current limitations to implementing value-based MEAs in Europe and potential ways forward; and the role of the patient community to enhance early access initiatives as mediators and decision-makers on national and European levels.
Highlights from the Panel Debate
Nicola Bedlington: Adaptive Pathways are of interest to patients as they offer the opportunity to address the bottlenecks evident in standard pathways. Patients have a role throughout the entire development lifecycle. Participation in early dialogues is crucial, given that patients have different perspectives on benefit/risk from the other stakeholders. Patients – and caregivers – are willing to accept greater risks, however not at any cost. We cannot be cavalier regarding safety and need to consider patient preferences for trade-offs between earlier access and potential risks. These preferences are not static or linear. Expectations and perceptions of benefits and risks change over time and according to age, illness, role in society, culture.
Patients equally have a role in defining the value of an innovation, in determining the significant added benefit of a novel treatment. Patient empowerment is thus part of the drive towards sustainability of our health systems.
“Adaptive Pathways (MAPPS), as defined by ADAPT SMART, foster access to beneficial treatment for the right patient groups at the earliest appropriate time in the product life–span in a sustainable fashion.” Nicola Bedlington
Ad Schuurman: Payers share the concerns regarding sustainability. The pace of biomedical innovation is higher than it has ever been before. These faster advances are good for patients, and we welcome the patient voice. But there needs to be a balance, some patient groups are very organised, however, we are also responsible for those patients that are not as vocal. It is our responsibility to have control over costs, and what you spend on one patient, you do not have for the other. The dynamics are so quick and the standard of care changes so rapidly, that we are at risk of losing control, not knowing what we are paying for in the hospital setting.
This is also connected to the ways we assess data and the quality of evidence, which is becoming quickly outdated in this highly dynamic environment. And we do not have new ways of assessing real-world data (RWD) in place in Europe. There is a lot of promise in real-world evidence, but we do not see it, cannot use it yet. So Adaptive Pathways are of interest to payers if we can regain a measure of control in the funding of innovations.
“Adaptive Pathways should be restricted to special cases, where there is a high unmet patient need, or an urgent public health requirement, and where we expect a major improvement over current options.” Ad Schuurman
Rob Thwaites: It has traditionally been industry’s role to generate data, and we should produce evidence on the unmet needs as well as the value a new compound is expected to bring in addressing the patient needs. This real-world evidence (RWE) complements the randomized clinical trial data and supports an iterative scientific development in Adaptive Pathways. Companies in the pharmaceutical industry have been introducing more systematic approaches for effectiveness planning, incorporating real-world research into the activities of the project teams for drugs in development. This includes engaging earlier in scientific advice processes, not only with regulators but also with HTA agencies and patient organisations, even where the range and disparity of evidence needs may well give rise to concerns that the manufacturer will not be able to meet all needs.
“Adaptive Pathways emphasize iterative development, permitting a stepwise approval in broadening populations, early dialogues with all stakeholders, and real-world evidence supplementing clinical trials.” Rob Thwaites
Rob: At the same time there are a number of areas of RWE that require further development, including the methodological approaches, the quality of the evidence, and the data sources. There is also the issue of acceptability of RWE, which differs between stakeholders and countries.
Nicola: This is a key issue, while stakeholders can generally agree on the unmet need, their evidence requirements differ substantially. Can we align regulatory with HTA/payer data needs, and develop a fair framework to define the value and the pricing of innovations, as well as a set of basic principles that all stakeholders can agree on? We should utilise the opportunities and all the work that is ongoing, for example within IMI’s Big Data for Better Outcomes (BD4BO) program.
Ad: There are many reasons for the differences between countries, and these will persist. Alignment is relatively unlikely. Payers are often not involved, and even though there is 80-90% overlap in data needs between HTAs and payers, priorities differ. Some countries will assess RWE, while others do not, and may never do so. Perhaps for orphan drugs there is a chance for greater consensus as there is a culture of international collaboration within the European rare disease networks.
“The IMI GetReal RWE Navigator is a user-friendly tool for all those considering effectiveness research, including methodological guidance and a directory of resources.” Rob Thwaites
Ad: Adaptive Pathways will require new and adaptive reimbursement strategies with mutually accepted prices. This could be managed entry agreements (MEAs) with conditional reimbursement at an initial level, which will be re-assessed once the data has matured, leading to a higher or lower price, or no reimbursement, if the initial promise of the innovation is not confirmed. This also includes realistic exit strategies which are clearly agreed upon in advance between all stakeholders. Especially patients and doctors need to be aware of this.
The main problem is that the payers do not have the resources to manage these MEAs in a proper way, which is why we must focus on the most urgent cases with the greatest unmet need. In addition, as mentioned, we do not have the structure to assess the outcomes data. This is why most of the current MEAs are financial- and not outcomes-based in Europe. We must therefore re-think the assessment of value of therapeutic innovations, focussed on the outcomes achieved.
“We need to overcome the old way of pricing and develop adaptive reimbursement models, paying less initially and then paying more when the data has matured, and real-world patient outcomes are clearer.” Ad Schuurman
Rob: Agreed, we still have a long way to go regarding value-based MEAs. We have seen from the Italian experience with these agreements that building the infrastructure to collect the necessary data is not straightforward. Is there an appetite for MEAs within senior levels of management in pharma? I think so, but we need to see positive examples of how MEAs could work.
Ad: Payers will need to push the issue by increasingly saying “no” to the traditional pricing models.
Nicola: The patients’ role in MEAs is to share their health outcomes data. Particularly those with serious and chronic conditions are willing to do so. Wider citizen groups are more critical, and we should make greater efforts to shift public opinion toward a more positive approach to research. EUPATI is doing this with a multi lingual toolbox on medicines R&D.
EUPATI has also educated about 100 patient-experts through an expert level course, and 60 more patients will graduate later this year. We are fostering patient engagement throughout the development life-cycle, with patient advisors as equal partners at the table with EMA, HTAs, payers and pharma.
There is much to do: massive challenges persist across Europe to equitable and timely access, not just to innovative medicines but also to the most basic care. This “post-code lottery” is unacceptable in the 21stCentury and we must address this. It’s a human rights issue, nothing more, nothing less.
“The vision is a fair framework and a set of basic principles to define the value and pricing of innovation, which all stake-holders can agree to.” Nicola Bedlington
References and Further Reading
- ADAPT SMART, Accelerated Development of Appropriate Patient Therapies a Sustainable, Multi-stakeholder Approach from Research to Treatment-outcomes > Home: http://adaptsmart.eu/home/
- IMI GetReal, Real-Life Data in Drug Development > Home: http://www.imi-getreal.eu
- EUPATI, European Patients' Academy, Patient Education > Home: https://www.eupati.eu
- Bedlington N, Patients and EU Health Policy: Setting the Scene, Presentation, The Hague, 6 June 2016, EPF Regional Advocacy Seminar, http://www.eu-patient.eu/globalassets/events/2016/2016-ras/nicola_bedlington_setting_the_scene_june_2016.pdf
- Schuurman A, Assessment for reimbursement: Collaboration at EU-level, the MEDEV-experiment, London School of Economics and Political Science, Volume 14 Number 3, 2008 http://www.euro.who.int/__data/assets/pdf_file/0016/80440/Eurohealth14_3.pdf
- Schuurman A, Has ADAPT SMART been successful in changing the way we think about approving new therapies? Interview, March, 2018, http://adaptsmart.eu/has-adapt-smart-been-successful-in-changing-the-way-we-think-about-approving-new-therapies-interview-with-ad-schuurman/
- Thwaites R (contributor): Advancing Evidence Generation for New Drugs IMI GetReal’s Recommendations on Real-World Evidence, IMI GetReal, 2017 (WP1 Deliverable) https://www.imi-getreal.eu/Portals/1/Documents/01%20deliverables/2017-03-29%20-%20WP1%20-%20Advancing%20Evidence%20Generation%20for%20New%20Drugs.pdf
- Gellad WF et al, Accelerated Approval and Expensive Drugs — A Challenging Combination, Perspective, N Engl J Med 376;21 nejm.org May 25, 2017, http://www.nejm.org/doi/full/10.1056/NEJMp1700446#t=article
- NEJM Perspective: Drug Regulation and Pricing — Can Regulators Influence Affordability? http://www.nejm.org/doi/full/10.1056/NEJMp1601294
- Frieden TR, Evidence for Health Decision Making — Beyond Randomized, Controlled Trials, N Engl J Med 2017;377:465-75. DOI: 10.1056/NEJMra1614394
- Rachel E et al, Real-World Evidence — What Is It and What Can It Tell Us? N Engl J Med 2016; 375:2293-2297 DOI: 10.1056/NEJMsb1609216
- Jarow JP et al, Multidimensional Evidence Generation and FDA Regulatory Decision Making, Defining and Using “Real-World” Data, JAMA August 22/29, 2017 Volume 318, Number 8, http://jamanetwork.com/journals/jama/article-abstract/2644655
- Duke Margolis Centre for Health Policy, A Framework for Regulatory use of Real-World Evidence, White Paper, September 13, 2017, https://healthpolicy.duke.edu/sites/default/files/atoms/files/rwe_white_paper_2017.09.06.pdf
- Gill J et al, The use of Real World Evidence in the European context: An analysis of key expert opinion, LSE Health, London School of Economics and Political Science, UK http://eprints.lse.ac.uk/68442/1/RWE_in_Europe_Paper1.pdf
- Anglemyer A et al, Healthcare outcomes assessed with observational study designs compared with those assessed in randomized trials. Cochrane Database of Systematic Reviews 2014, Issue 4. Art. No.: MR000034. DOI: 10.1002/14651858.MR000034.pub2.
- Makady A et al, Policies for Use of Real-World Data in Health Technology Assessment (HTA): A Comparative Study of Six HTA Agencies, Value in Health 20 (2017) 520 – 532 http://dx.doi.org/10.1016/j.jval.2016.12.003
- An Interview with Hans Georg Eichler, Senior Medical Officer at EMA, “Adapt or Die: Real-World Evidence Across the Lifecycle”, http://social.eyeforpharma.com/market-access/adapt-or-die-real-world-evidence-across-lifecycle
- Martinalbo et al, Early market access of cancer drugs in the EU, Annals of Oncology 27: 96–105, 2016, doi:10.1093/annonc/mdv506
- EURORDIS Call on Payers, http://www.eurordis.org/sites/default/files/call-on-payers.pdf
- Mechanism of Coordinated Access to orphan medicinal products (MoCA), http://www.eurordis.org/content/moca
- Wilson WH, A company experience of the first MoCA pilot project, Orphanet J Rare Dis. 2014; 9 (Suppl 1): O26, Published online 2014 Nov 11. doi: 10.1186/1750-1172-9-S1-O26
- EMA Parallel consultation with regulators and health technology assessment bodies http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001857.jsp&mid=WC0b01ac0580a11c96


PART 2: Timely Patient Access to Transformative Medicines: Early Access VALUE
PART 1: Timely Patient Access to Transformative Medicines: Early Access STRATEGY
Strasburger CJ, Karavitaki N, Störmann S, Trainer PJ, Kreitschmann-Andermahr I, Droste M, Korbonits M, Feldmann B, Zopf K, Fazal-Sanderson V, Schwicker D, Gelbaum D, Haviv A, Bidlingmaier M, Biermasz NR, Patient Reported Outcomes of Parenteral Somatostatin Analogue Injections in 195 Patients with Acromegaly, Eur J Endocrinol March 1, 2016 174 355-362, doi: 10.1530/EJE-15-1042
http://www.eje-online.org/content/174/3/355
Abstract
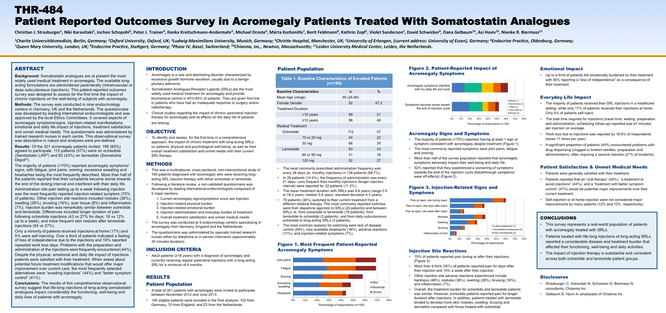
Background Long-acting somatostatin analogues delivered parenterally are the most widely used medical treatment in acromegaly. This patient-reported outcomes survey was designed to assess the impact of chronic injections on subjects with acromegaly.
Methods The survey was conducted in nine pituitary centres in Germany, UK and The Netherlands. The questionnaire was developed by endocrinologists and covered aspects of acromegaly symptoms, injection-related manifestations, emotional and daily life impact, treatment satisfaction and unmet medical needs.
Results In total, 195 patients participated, of which 112 (57%) were on octreotide (Sandostatin LAR) and 83 (43%) on lanreotide (Somatuline Depot). The majority (>70%) of patients reported acromegaly symptoms despite treatment. A total of 52% of patients reported that their symptoms worsen towards the end of the dosing interval. Administration site pain lasting up to a week following injection was the most frequently reported injection-related symptom (70% of patients). Other injection site reactions included nodules (38%), swelling (28%), bruising (16%), scar tissue (8%) and inflammation (7%). Injection burden was similar between octreotide and lanreotide. Only a minority of patients received injections at home (17%) and 5% were self-injecting. Over a third of patients indicated a feeling of loss of independence due to the injections, and 16% reported repeated work loss days. Despite the physical, emotional and daily life impact of injections, patients were satisfied with their treatment, yet reported that modifications that would offer major improvement over current care would be ‘avoiding injections’ and ‘better symptom control’.
Conclusion Lifelong injections of long-acting somatostatin analogues have significant burden on the functioning, well-being and daily lives of patients with acromegaly.
Strasburger CJ, Karavitaki N, Schophol J, Trainer PJ, Kreitschmann-Andermahr I, Droste M, Korbonits M, Feldmann B, Zopf K, Fazal-Sanderson V, Schwicker D, Gelbaum D, Haviv A, Biermasz
NR, Patient Reported Outcomes Survey in Acromegaly Patients Treated with Somatostatin Analogues, Poster presented at ENDO 2015, 97th Annual Meeting of the Endocrine Society, San Diego,
March 2015
Timothy Juday, PhD, Steven Fosburg, MA, Neal S. Mantick, MS, M. Haim Erder, PhD and David Schwicker, MA. Design of a registry of hypertension patients treated with β-blockers in Germany and the United States. Abstract and Presentation, ACCP Annual Meeting (October 14–17, 2007), Denver, CO

Schwicker D, Banz K, Roferon-A in Chronic Viral Hepatitis: Treatment - Clinical Outcomes - Cost-Effectiveness, Peter Lang, Berne, Basle, 115 pages, 1994
Chronic liver infection due to viral hepatitis B and C is a major public health problem. Worldwide, an estimated 750 million people are carriers of these two viruses. Alpha-interferons, such as
Roferon-A, are currently the therapy of choice in chronic viral hepatitis B and C. They are effective in preventing disease progression and life-threatening sequelae: liver cirrhosis, ascites,
hepatic encephalopathy, and variceal hemorrhage and/or hepatocellular carcinoma. On the other hand, discussion has been raised on the potentially enormous cost impact of these genetically
engineered drugs in the treatment of an already costly disease. This study identifies and evaluates the long-term clinical and economic effects of Roferon-A treatment in chronic viral hepatitis B
and C over a time period of 10 to 20 years. Clinical and epidemiological data, as well as the detailed health care resource use and cost assessment, reflect the clinical and economic reality in
Germany in 1993. A Markov model is used to predict the long-term clinical benefits of Roferon-A therapy in terms of reduced morbidity and mortality. Based on this, direct costs, savings, and
cost-effectiveness are analysed, including: Roferon-A therapy initiation and follow-up, physician services, laboratory services, medicotechnical services, drug therapy, hospitalization as well as
surgical interventions (liver transplantations).